Although signature analysis has been instrumental and transformed our understanding of mechanisms of genomic instability in cancer, there are still several important shortcomings in the current statistical modelling approaches. Addressing these challenges require advanced computational methods that can incorporate the complexity of mutagenesis in cancer. Our lab contributes to the development of the next-generation of signature analysis methods by rethinking the design of the models we use.
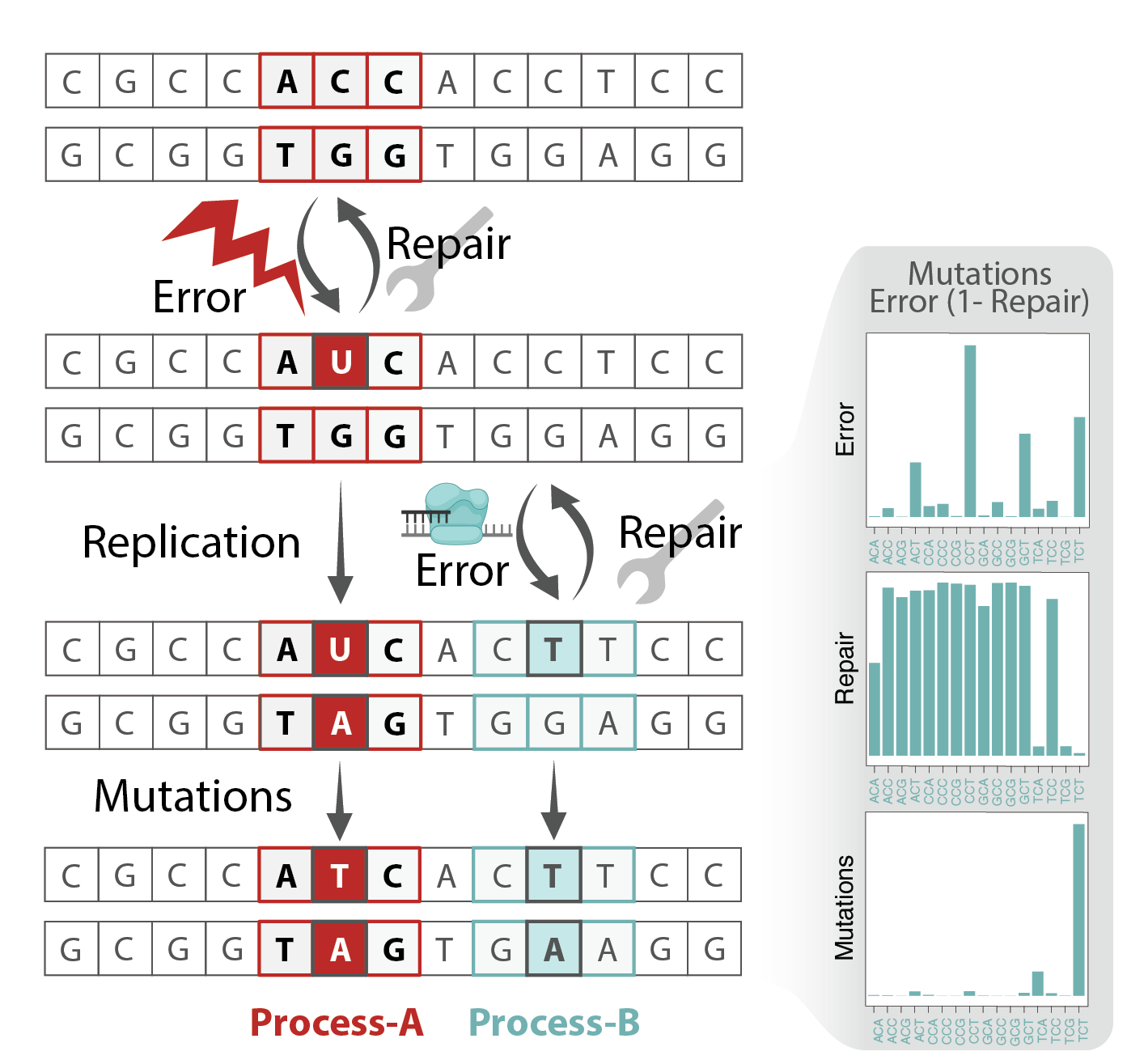
An important consideration is the dependence of signatures across genomic loci and time (see next section). Activitiy of each process depends on various features such as replication/transcription timing and strand, histone marks and genome organization. A big contributor to the variability is the repair pathways activity of which are highly influenced by the epigenome, replication, transcription and cell cycle. Relying on the vast history of research on these links and combining it with the findings from loci dependent signature analysis enable improved mechanistic interpretation and help in annotation of etiology of signatures. Additionally, these methods are instrumental in determining how signatures contribute to key cancer-promoting events.